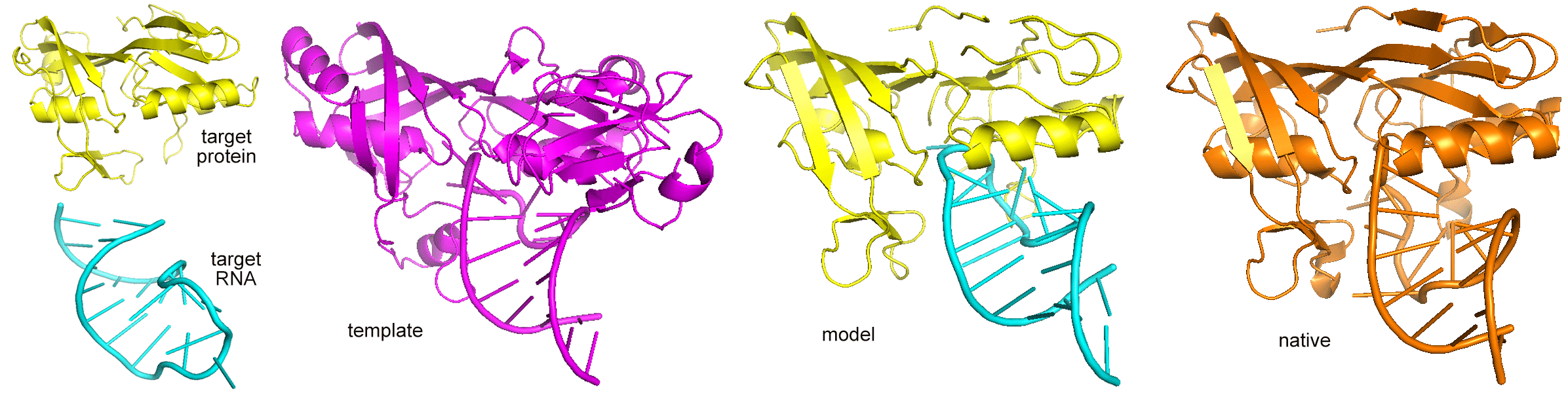
PRIME 2.0 is a Protein-RNA Interaction ModElling
program ,which includes TMalign (protein structural alignment) and
RMalign(RNA structural alignment) for searching template in the library. Protein
and RNA structure coordinates are needed. The output is the protein RNA-complex structure model.
The similar score (TMscore or complex structural score),which describle the binding similarity
between the target and the template, and a transformation matrix are also reported.
PRIME2.0 is available.